Clinical investigations for medical devices
How to notify the MHRA of your intention to carry out a clinical investigation for medical devices.
You may need to carry out a clinical investigation as part of the process to obtain a UKCA, CE or CE UKNI marking for your medical device. You must inform the MHRA if you are planning to do this at least 60 days before starting your investigation.
For studies in Great Britain, use this flow chart to decide if you are required to submit a formal clinical investigation.
For studies in Northern Ireland or Great Britain and Northern Ireland, use the flow chart and accompanying guidance in the Northern Ireland section below.
Note: applications under Annex XVI of the Medical Device Regulation (EU) 2017/745 (MDR) for devices with no intended medical purpose cannot be accepted in GB.
Clinical investigation enquiries
For specific enquires relating to applications for clinical investigations, contact info@mhra.gov.uk and ensure you add ‘clinical investigation enquiry’ in the subject line.
MHRA guidance
Follow the guidance on compiling a submission and when preparing your notification application.
Applications are submitted electronically using the Integrated Research Application System (IRAS).
See information for clinical investigators for what is required by clinicians involved in the investigation.
Check the electrical guidance for clinical investigations.
Check the information on the for the scientific data you must submit.
Check ) for presenting statistical information for your clinical investigation.
See the guidance on UKCA markings.
Regulatory advice meetings
The MHRA clinical investigations team can offer a comprehensive device regulatory advice meeting.
We cannot review individual documents but we can provide guidance on navigating the regulatory landscape.
See our information on fees regarding the charges for a regulatory advice meeting
To request a regulatory advice meeting, contact the Head of Clinical Investigations mark.grumbridge@mhra.gov.uk.
Validation checklist
When we receive your application for a clinical investigation of a medical device our regulatory handers will validate your application against the Clinical investigation application checklist. Use this checklist to help you submit a valid application.
How to notify the MHRA of your clinical investigation
Start your application via IRAS
Note: a notification to the MHRA will not be required for medical devices that are UKCA, CE or CE UKNI marked for the purpose that is under investigation.
Fees
See See section section 9 of our fees page.
A fee under the Medical Devices Regulations 2002:
(a) shall be payable when the notice to which it relates is given to the Secretary of State;
and
(b) shall accompany that notice when it is given.given
Payment for a clinical investigation
To enable accurate and timely invoicing, you should provide the following information when applying for your clinical investigation:
- company name and full address
- contact name and email address of the individual responsible for paying the fee
- a company invoice or transaction number (if applicable)
Once you have received your invoice and made the payment, you should send evidence of payment by email to CI-applications@mhra.gov.uk, quoting the MHRA reference (CI/XXXX/XXX).
In applying the regulation and to avoid delays, you must pay the the MHRA upon confirmation of a valid application for a clinical investigation or amendment to a clinical investigation.
We will not provide a final regulatory decision (approval or objection letter) on the application or amendment until we have received payment.
Payment easements for small,a andclinical medium enterprisesinvestigation
- Easements
ToareenableavailableaccuratetoandSMEtimelyforinvoicing,clinical investigations. At the point of submission of the application you mustshouldprovide the MHRAfollowingSMEinformationapprovalwhenletterapplyingfor theyoureasementclinicalto be considered. - Upon
companyreceiptnameof a validation of an application MHRA will request 50% of the fee and thefullremaining 50% will be payable within six months after the first invoice has been issued.address - If
contactthenamefinaland50%emailaddressindividualfeeresponsibleis not paid, then any approval letter for apayingclinical investigation provided by MHRA will be revoked resulting in the clinical investigation being stopped.fee - In
athecompanycaseinvoiceof an objection or atransactionwithdrawalnumberof(ifanapplicable) - No
andeasementmadeistheofferedpayment,foryoudeviceshouldregulatorysendadviceevidencemeetings - For
offurtherpaymentinformation,byseeemailPaymenttoeasementsCI-applications@mhra.gov.uk,andquotingwaiverstheforMHRAsmallreferenceand medium companies
Once applicationyou thehave remainingreceived feeyour isinvoice due
Assessment
This section concerns clinical investigations being conducted in Great Britain only. See the separate section below for studies involving Northern Ireland.
When we have received your documents and validated them, we will contact to you within 5 working days to confirm that the 60-day assessment period has started, or we will let you know if there are any issues. If we raise any issues, the 60-day assessment period will start when we receive a valid response.
Day 1 of the 60 days is taken as being the first day that follows the date of acceptance of a valid application. For example, if an application is received on 24 August and the assessor validates the submission on 28 August, the clock starts on 29 August.
During the assessment, experts will assess the safety and performance of your device as well as the design of the clinical investigation to be carried out. We will write to you if we require further information. It is essential that you contact us as soon as possible if you require clarification.
If there are possible grounds for objection, where possible we will arrange a teleconference for a better understanding and to find a resolution within the 60-day assessment time.
We will send you a letter by the 60th day with a decision (‘objection’ or ‘no objection’) whether or not you can carry out the proposed clinical investigation.
Serious adverse event (SAE) reporting
This section concerns clinical investigations with sites in Great Britain, and clinical investigations with sites in both Great Britain and Northern Ireland. Please see the separate section below for studies with Northern Ireland sites only (no sites in Great Britain).
In line with UK Regulations and MEDDEV 2.7/3, all reportable events must be fully recorded and notified to the MHRA. This includes all serious adverse events, irrespective of whether the device has been assessed as having a causal relationship, and reportable events (per MEDDEV 2.7/3) occurring in third countries in which a clinical investigation is performed under the same clinical investigation plan. These can be provided using either the MEDDEV 2.7/3 SAE reporting table, or the MDCG 2020-10/2 SAE reporting table, as long as the required information is included.
Please submit an SAE reporting form in the new MORE portal with your completed table attached.
See details on how to register for the MORE portal.
MEDDEV 2.7/3 also contains further guidance on clinical investigation reporting in Great Britain.
Quarterly summary reports
As a condition of MHRA approval for a clinical investigation, in addition reporting individual serious adverse events as detailed above, you must send us quarterly summary reports providing an update on the latest overall safety profile for the investigation.
To provide these summaries, use the . This template is for devices only but you should also use it for device-related reporting on any combined studies. Do not include detail on any investigational medicinal product (IMP) under investigation.
Submit your quarterly summary reports directly through https://more.mhra.gov.uk/login
End of study reports
Email your end of study report to CI-applications@mhra.gov.uk.
Study deviations
Sponsors must notify the MHRA of all deviations (relating to UK study sites only) as soon as they are aware of them. Include details about the nature of the deviation, when it occurred, where it occurred, and any proposed corrective and preventative actions.
Use the following MHRA protocol deviation tracker Excel template when reporting deviations and keep this as a ‘live’ document so that new deviations can be added. This enables both the sponsor and the MHRA to have a complete overview each time it is submitted.
Send the completed spreadsheet by email to info@mhra.gov.uk.
Combined review of a CTIMP and medical device
Combined review is the way research teams request approval for clinical trials of investigational medicinal products (CTIMPs) and combined medicine and device trials.
Research teams make a single application using a new part of IRAS, which goes to both the MHRA and a research ethics committee (REC) at the same time. The application also goes for study wide review, such as HRA and HCRW approval, if the study is to take place in the NHS or Northern Ireland HSC.
The regulatory and ethics reviews take place in parallel and any requests for further information (RFIs) are raised jointly. A single response to these requests leads to a single decision from both reviews. Study-wide review is usually issued at the same time as MHRA and REC, but may come later if there are still issues to discuss with the applicant.
IRAS is used for the initial application and supports the trial through amendments right up to the end of the trial. Research teams can allocate different roles in the system for colleagues working on the project.
Further information is available in our Combined IMP device guidance.
Amendments
This section concerns clinical investigations conducted in Great Britain only. See the separate section below for studies involving Northern Ireland.
Once you’ve received a letter of no objection from us, you must notify us of all proposed amendments to the investigation. You must wait until we send you another letter of no objection before you make the changes.
You must tell us about any changes made to:
- the device under investigation
- study documentation, including the clinical investigation plan
- investigators or investigating institutions
- changes requested by an ethics committee
If you do not tell us about proposed amendments, you could be liable to prosecution.
Begin your amendment submission on on HRA IRAS.
Refer to the the IRAS user user guide’s section on amendments.
When you notify the the MHRA of of amendments, include the following:
- a covering letter with the MHRA reference number for the clinical investigation (for example, CI/2023/XXXX)
-makesureyouincludethefollowingdetailsforthepersonmakingtheamendmentpayment:companynameandfulladdresscontactnameandemailaddressoftheindividualresponsibleforpayingthefeeifapplicableaPOnumberortransactionnumber
- a table with a summary of each proposed change with the justification for each change
- red-lined (showing changes being made) and clean copies of all amended study documentation
- a signed statement by, or on behalf of, the manufacturer that the proposed change(s) do not predictably increase the risk to the patient, user or third party
proofofpayment(followthepaymentprocessasoutlinedinthesectionbelow)
If you do not follow the guidance above, the submission will be returned to you.
Email the amendments to CI-amendments@mhra.gov.uk, unless the files are too big, in which case contact us at the same email address to request a link for uploading the documents. We will only process amendments submitted to this mailbox. Do not submit amendments to individuals or any other mailbox.
Payment for an amendment
UnderYou will need to pay a fee for amendments to clinical investigations (see Fees above).
Note: we do not consider an amendment submission valid until we have received all required documents (including proof of payment). We will not review the newamendment feesor implementedprovide ina Julyfinal 2025regulatory theredecision is(approval noor feeobjection payableletter) foruntil anythis time.
To make a payment:
MakeapaymentviaMakeapaymenttoMHRAquotingtheMHRAreference(CI/XXXX/XXXX)andthenumber(forexample,amendmentNo1)astheinvoicenumber.Onceyouhavemadethepayment,submityouramendmentCI-amendments@mhra.gov.ukclinicalincludinginvestigation.allthedocumentsrequiredasstatedabove.
Ensure that you provide evidence of payment and quote the MHRA reference number (CI/XXXX/XXX) and amendment number as the email subject.
After you have received the decision letter for your amendment, you will receive an official invoice. This is for your records only and no further action is required.
Early termination or temporary halt of a clinical investigation
Sponsors must notify us of the early termination of a clinical investigation and provide a justification for the early termination as stated in Regulations 16(11) and Section 29(10) of the UK MDR 2002.
Send a copy of the final written report of a clinical investigation of a device falling within the scope of the UK MDR 2002 (Regulations 16(10) and 29(9)) to CI-applications@mhra.gov.uk.
Sponsors should also notify us of a temporary halt of a clinical investigation.
Send the notifications to info@mhra.gov.uk.
Northern Ireland
The Northern Ireland Protocol requires Northern Ireland to continue to align with EU rules for devices after 1 January 2021. Therefore, the Medical Device Regulation (EU) 2017/745 (MDR) and the in vitro Diagnostic Medical Device Regulation (EU) 2017/746 (IVDR) will apply in Northern Ireland from 26 May 2021, and 26 May 2022 respectively, in line with the EU’s implementation timeline.
All clinical investigations requiring application to the MHRA (which involve a site in Northern Ireland) must be submitted to the MHRA in line with the requirements of EU MDR 2017/745.
Note: this single application will also cover any sites proposed in Great Britain in addition to site(s) in Northern Ireland for the same clinical investigation.
For clinical investigations involving a site in Northern Ireland, use this flow chart and accompanying guidance to determine if you need to submit an application to the MHRA.
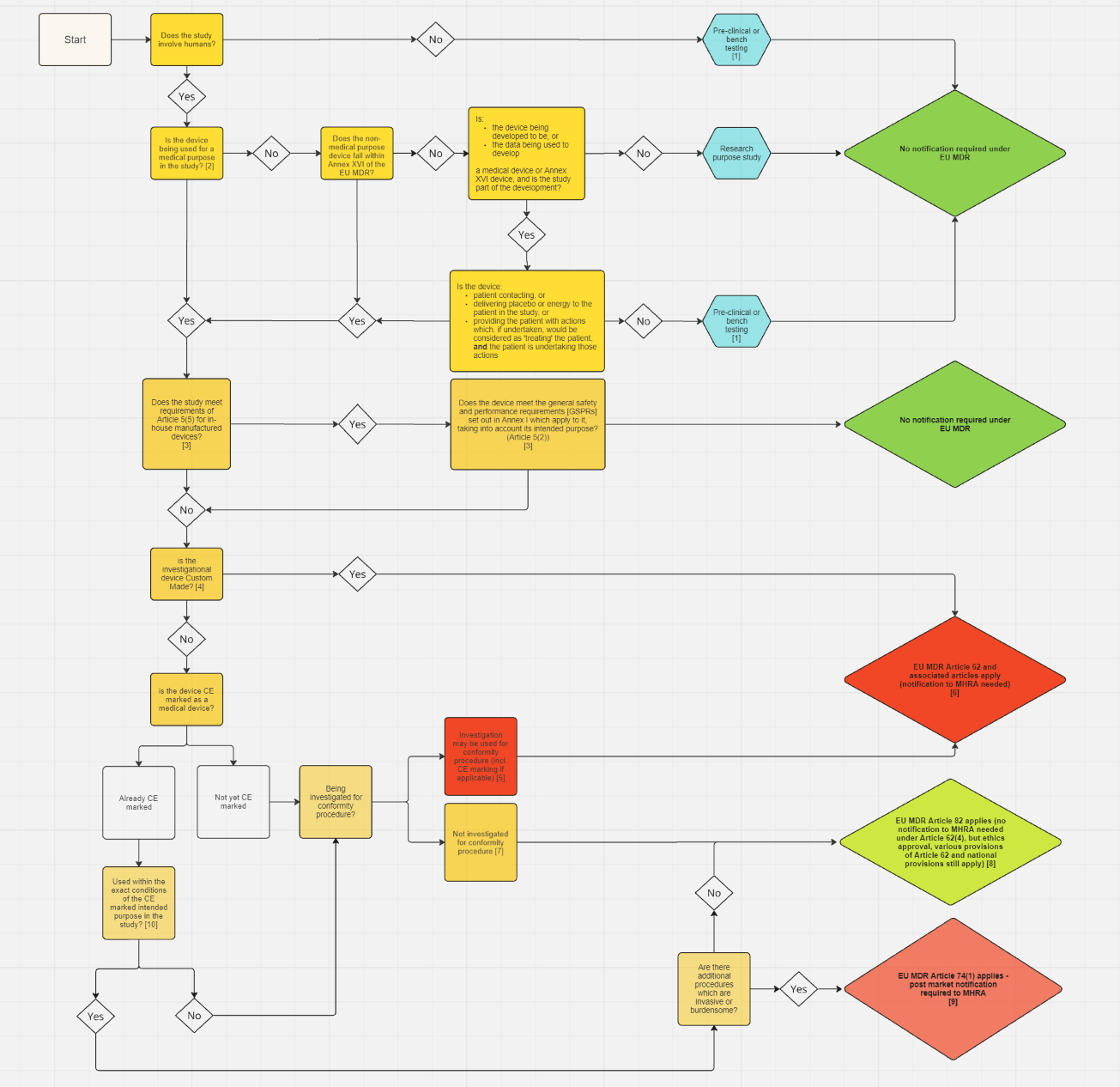{kind=link}
The following sections provide guidance for these applications and the requirements for amendments and post market studies involving sites in Northern Ireland.
Assessment of CI applications
When we have received your documents and validated them, we will write to you within 10 calendar days to confirm that the application is valid and the assessment has started, or we will let you know if there are any issues. If there are any issues, we will confirm these in writing and provide a 10 calendar day deadline for a response. The assessment will not start until we have received a valid response. If, after receipt of the response, we still consider the application invalid, or the 10 day deadline has expired, we will write to confirm this within 5 calendar days.
Day 1 of the MHRA assessment is taken as being the date that we confirm that we have received a valid application. During the assessment, experts will assess the safety and performance of your device as well as the design of the clinical investigation to be carried out.
We will write to you if we need further information. It is essential that you contact us as soon as possible if you need clarification. If there are possible grounds for refusing authorisation, where possible, we will arrange a teleconference for a better understanding and to find a resolution within the assessment period.
Depending on whether we consult experts, we will inform you of our decision within 45 or 65 calendar days as per the EU MDR. This period will be suspended if we request additional information for a maximum of 7 calendar days for each request, up to a maximum of 3 requests. Any further requests will not result in a clock stop.
We will send you a letter by the final day (or before) with a decision whether or not you can carry out the proposed clinical investigation.
Serious Adverse Event (SAE) Reporting under Regulation (EU) 2017/745
This section concerns clinical investigations with sites in Northern Ireland (but no sites in Great Britain). For studies with sites in Great Britain, or for studies conducted in both Great Britain and Northern Ireland, please see the separate section above.
The reporting requirements of Regulation (EU) 2017/745 apply to these studies.
The sponsor shall fully record all of the following:
- any adverse event of a type identified in the clinical investigation plan as being critical to the evaluation of the results of that clinical investigation;
- any serious adverse event;
- any device deficiency that might have led to a serious adverse event if appropriate action had not been taken, intervention had not occurred, or circumstances had been less fortunate;
- any new findings in relation to any event referred to in points (a) to (c).
The sponsor shall report, without delay to MHRA, all the following:
- any serious adverse event that has a causal relationship with the investigational device, the comparator or the investigation procedure or where such causal relationship is reasonably possible;
- any device deficiency that might have led to a serious adverse event if appropriate action had not been taken, intervention had not occurred, or circumstances had been less fortunate;
- any new findings in relation to any event referred to in points (a) and (b).
Reports can be provided using the MDCG 2020-10/2 SAE reporting table.
Please submit an SAE reporting form in the MORE portal with your completed table attached.
See details on how to register for the MORE portal.
MDCG 2020-10 also contains further guidance on clinical investigation reporting for Northern Ireland.
For enquiries please see the above section of this guidance page.
Amendments/modifications
Once you’ve received authorisation from us to conduct the clinical investigation in Northern Ireland, you must notify us of all proposed modifications to the investigation before you make any changes. You must wait until we send you another letter of authorisation before you make substantial modifications. Depending on whether we consult experts, we will send you our decision within 38 or 45 calendar days.
You must tell us about all modifications, but only those considered to be substantial will require authorisation by the MHRA.
Substantial modification includes:
- changes to the medical device under investigation
- changes to the design or methodology of the clinical investigation, or to background information
- changes to the procedures undertaken by participants
- changes to the risk/benefit assessment for the study
- significant changes to study documentation such as clinical investigation plan, investigator’s brochure, participant information sheets, consent forms, letters to GPs or other clinicians, information sheets for relatives or carers
- appointment of a new chief investigator
- inclusion of a new trial site (not listed in the original application)
- appointment of a new principal investigator at a trial site
- planned
temporaryrestarthaltof a study aftertoaprotecttemporaryparticipantshalt,fromifharm,changesandhavethebeenplannedmaderestarttooftheadevicestudyorfollowingstudyatemporaryhalt - a change to the definition of the end of the study
- extension of the study beyond the period specified in the application form
- any other significant change to the protocol
- changes requested by an ethics committee
For non-substantial modifications you only need to notify us to ensure our records are up to date. If upon review of the proposed non-substantial modification, we consider it to be in the substantial category, we will inform the sponsor, and the proposed modification should not take place until an MHRA authorisation is received.
Non-substantial modifications are changes that are unlikely to have a substantial impact on the safety, health or rights of the subjects or on the robustness or reliability of the clinical data generated by the investigation and include:
- a change of sponsor(s) or sponsor’s legal representative
- a change to the insurance or indemnity arrangements for the study
- minor changes to the protocol or other study documentation, e.g. correcting typographical errors, updating contact points, minor clarifications
- changes to the chief investigator’s research team
- changes to the research team at particular trial sites (other than appointment of a new principal investigator)
- changes in funding arrangements
- minor changes in the documentation used by the research team for recording study data
- changes in the logistical arrangements for storing or transporting samples
Post market studies
You must notify us of all clinical investigations conducted in Northern Ireland involving CE marked devices that also involve procedures additional to the normal conditions of use of the device, that are also invasive or burdensome.
You must make the notification at least 30 days before starting the study.
When you notify us of these clinical investigations, we need the following information:
- application form
- investigator’s brochure
- clinical investigation plan or protocol
- signed statement
- REC opinion
- proof of insurance cover or indemnification of subjects
- PIS and IC
- arrangement to ensure protection and confidentiality of personal data
- details of the technical documentation (risk analysis, test reports) kept available
Submit your application electronically using the Integrated Research Application System (IRAS).
Early termination or temporary halt of clinical investigation
Sponsors must notify the MHRA of the temporary halt or early termination of a clinical investigation and provide a justification within 15 days, or 24 hours if the decision was taken on safety grounds. The clinical investigation report and summary must be provided within 3 months for such studies that are temporarily halted or terminated early.
Medical devices: clinical investigations and performance studies in Northern Ireland
Information about changes to the operation of certain clinical investigations and performance studies in Northern Ireland.
Certain clinical investigations and performance studies that take place in Northern Ireland require the sponsors to be established either in Northern Ireland or the EU or to have a legal representative established in Northern Ireland or the EU.
The rules that apply (Medical Device Regulation (Regulation 2017/745) (MDR) and In Vitro Diagnostics Regulation (Regulation 2017/746) (IVDR) allow the UK to disapply these rules.
This requirement applies if the investigation or study is only taking place in its territory and not in the EU. Therefore, the MHRA is disapplying the requirement for the sponsor or their legal representative of a clinical investigation or performance study to be established in Northern Ireland or the EU, provided all of the following conditions are met:
- the investigation or study must also be taking place in both Northern Ireland and Great Britain
- the investigation or study must not be taking place in an EU member state
- the sponsor must be either established in or have a written agreement with a legal representative in Great Britain, who is responsible for ensuring compliance with the sponsor’s obligations in the MDR or IVDR
- the sponsor must establish a contact person in Northern Ireland for the clinical investigation or performance study, who will be the addressee for all communications with the sponsor provided for in MDR or IVDR
- any communication with that contact person is deemed to be communication with the sponsor
All remaining requirements under the MDR and IVDR apply. This guidance will take immediate effect.
For further information, contact the MHRA info@mhra.gov.uk.
In vitro diagnostic medical devices (IVDs)
Unless an exemption applies, all IVD devices being placed on the market or put into service in the UK must have the relevant mark of conformity (UKCA, CE or UKNI).
This includes IVD devices used in clinical trials of medicines (CTIMPs) to stratify patients for inclusion/exclusion in the trial or stratified to a cohort in a trial.
At the time of the clinical trial application, where clinical performance of the IVD device is yet to be demonstrated, for CTIMPs taking place in GB, the IVD device must have demonstrated evidence supplied to the MHRA for the analytical performance of the IVD (for example, detection of a biomarker) as summary reports of each study or protocol. This will include reagents, equipment, calibrators, controls and software. For use of IVD devices in CTIMPs conducted in Northern Ireland, refer to guidance available on Clinical investigations and performance studies in Northern Ireland.
Alternatively, if the analytical performance study has not taken place, the sponsor must provide the MHRA with a tabular summary (MS Word Document, 49.7 KB) description of the analytical methods, including acceptance limits and parameters for performing validation. You should supply the contents requested without modifying the template structure.
Trials which determine the clinical performance of the assay must be registered as IVD performance evaluation studies.
See also guidance on in vitro diagnostic medical devices: guidance on legislation.
Special circumstances for healthcare establishments
You do not need to notify us of a clinical investigation if:
- you have manufactured the medical device in-house for your own patients with no objective to place it on the market
You may need to notify the MHRA of a clinical investigation if:
- you want to provide a medical device to another organisation, that up until now has been manufactured in-house for patients, for data to support safety and performance of a commercial product
See which may be relevant to you.
Health Research Authority (HRA) and Health and Care Research Wales (HCRW) Approval
HRA and HCRW approval applies to all project-based research taking place in the NHS in England and Wales. It brings together the assessment of governance and legal compliance, undertaken by dedicated HRA and HCRW staff, with the independent Research Ethics Committee (REC) opinion provided through the UK Research Ethics Service.
It replaces the need for local checks of legal compliance and related matters by each participating organisation in England and Wales. For information on how to prepare and submit an application for HRA and HCRW Approval, refer to the HRA website.
More information
For more details on classifications, see MEDDEV 2.4/1 for guidance on classifications.
If you have any questions before submitting your notification, email info@mhra.gov.uk.
Updates to this page
-
Revised wording in the 'Payment for a clinical investigation' section to remove reference to 'amendments to a clinical investigation'.
-
Added link to updated SME information (relating to fees).
-
Updated 'Amendments' and 'Fees' sections to reflect new fees applicable from 16 July 2025.
Update history
2026-07-06 15:54
Updated information: Applications for an investigative medicinal product and medical device – Parallel Review
2026-05-19 11:59
Notes added to section ‘Fee waiver programme details’
2026-05-14 15:58
Updated the section ‘Applications for an investigative medicinal product and medical device – IMP+Device’ with latest guidance
2026-05-06 12:56
Removed survey link from further information section as the survey has now closed.
2026-04-28 11:49
Added fee waiver cover letter requirement for a brief summary of the planned clinical investigation
2026-04-20 00:01
Updated the section Pilot of a medical device clinical investigation fee waiver programme for micro and small sized enterprises to add information on the fees waiver pilot
2026-04-09 15:48
Updated to fix broken link to Statistical Considerations PDF
2026-04-01 15:40
Updated to reflect the Pilot of a medical device clinical investigation fee waiver programme for micro and small sized enterprises has now closed
2026-01-14 15:00
Updated to add latest version of QSR Template Guidance
2026-01-12 14:07
Updated documents: MP+Device Review Applications for an investigational medicinal product and investigational medical device IMP+Device review process flowchart
2026-01-08 10:45
Updated changes consist of: • A re-order of the full webpage for ease of use• Updates to headings and corresponding contents page• Update to guidance on healthcare establishments, payments for clinical investigations and quarterly summary reports• Name change and new guidance on investigational medicinal products and device reviews• Link added to CTIMP guidance
2025-12-05 12:47
Update to ‘Assessment of CI applications’ wording
2025-11-24 16:31
Updates to Flow Chart for GB Clinical Investigations to greater align with UK MDRUpdates/expansion of GB Flow Chart Accompanying Guidance, with examplesAdded link to Northern Ireland performance studies submission guidanceRe-linking of CI biological safety guidance
2025-11-12 16:00
New section ‘Pilot of a medical device clinical investigation fee waiver programme for micro and small sized enterprises’.
2025-10-17 12:34
Updated to include link to combined medicine and device trials guidance.
2025-10-06 13:08
Updated file ‘Clinical investigations of medical devices – guidance for manufacturers’ to reflect the following changes:Clarified EU MDR can be met for GB only studies (page 4)Updated point 5 on when application to MHRA is required and added links on where to find MHRA flow charts (page 6)Some formatting fixes
2025-10-01 11:35
Major update to guidance wording
2025-09-26 12:33
Changed to add updated guidance on MFRs on clinical investigations jm
2025-09-05 15:43
Removal of the in vitro diagnostic medical devices (IVDs) guidance
2025-08-27 11:18
Added new flow chart and guidance documentation for UK and Northern Ireland.
2025-08-05 16:42
Revised wording in the ‘Payment for a clinical investigation’ section to remove reference to ‘amendments to a clinical investigation’.
2025-07-16 14:44
Added link to updated SME information (relating to fees).
2025-07-16 00:01
Updated ‘Amendments’ and ‘Fees’ sections to reflect new fees applicable from 16 July 2025.
2025-04-10 14:23
Updated to add new guidance for manufacturers document.
2025-02-20 16:52
Changed text for optimisation in both ‘Serious adverse event (SAE) reporting’ sections
2024-12-17 17:09
Updates to section ‘Northern Ireland’. Includes attachments for ‘flow chart’ and ‘accompanying guidance’ and revised wording underneath SAE reporting.
2024-12-04 09:51
Information about serious adverse event (SAE) has been updated.
2024-10-25 14:48
Added link to ‘IRAS User Guide – Amendments’.
2024-09-26 09:34
Updated section on ‘Amendments’ to reflect changes to the process.
2024-09-06 08:55
Updated ‘In Vitro Diagnostic Medical Devices (IVDs)’ section,
2024-08-27 11:09
Updated to include new QSR template
2024-08-05 11:45
New section ‘Regulatory advice meetings’ added to this page.
2024-07-02 16:17
Updates to clarify the fees and payment process, addition of guidance on early terminations and temporary halts in GB and NI and clarification that Annex XVI applications cannot be accepted in GB.
2024-04-26 15:15
Updated guidance for manufacturers PDF attachment
2024-04-23 15:43
Added links to: – guidance for manufacturers on clinical investigations- information for clinical investigators
2024-04-10 10:48
Added Combined review of a CTIMP and Medical Device section and accompanying guidance Combined IMP Device guidance
2024-03-20 16:27
‘Electrical guidance for clinical investigations’ has been uploaded
2024-03-14 15:35
Updated to add hyperlink on submission guidance
2024-01-16 10:21
Added template for submitting Quarterly Summary Reports.
2023-12-21 12:21
Added a PDF on our electrical guidance for clinical investigations. Added information to the ‘Amendments’ section.
2023-12-14 13:50
Added document ‘Tabular Summary input (template) for non marked IVD devices’.
2023-08-24 13:45
Added information on fees payable in relation to clinical investigation and amendments to clinical investigations.
2023-06-07 10:29
Updated information on ‘MHRA / HRA Coordinated pathway’ – Resuming Monday 22 May 2023.
2023-05-11 10:43
Added Validation Checklist text and document
2023-01-20 16:29
Updated information on how to submit the SAE reporting form.
2022-11-21 12:45
Clinical investigation numbers for 2021 have been included.
2022-09-21 09:27
Added section ‘Quarterly Summary Reports’
2022-05-19 17:05
Added section for “Study deviations” and related XLS.